

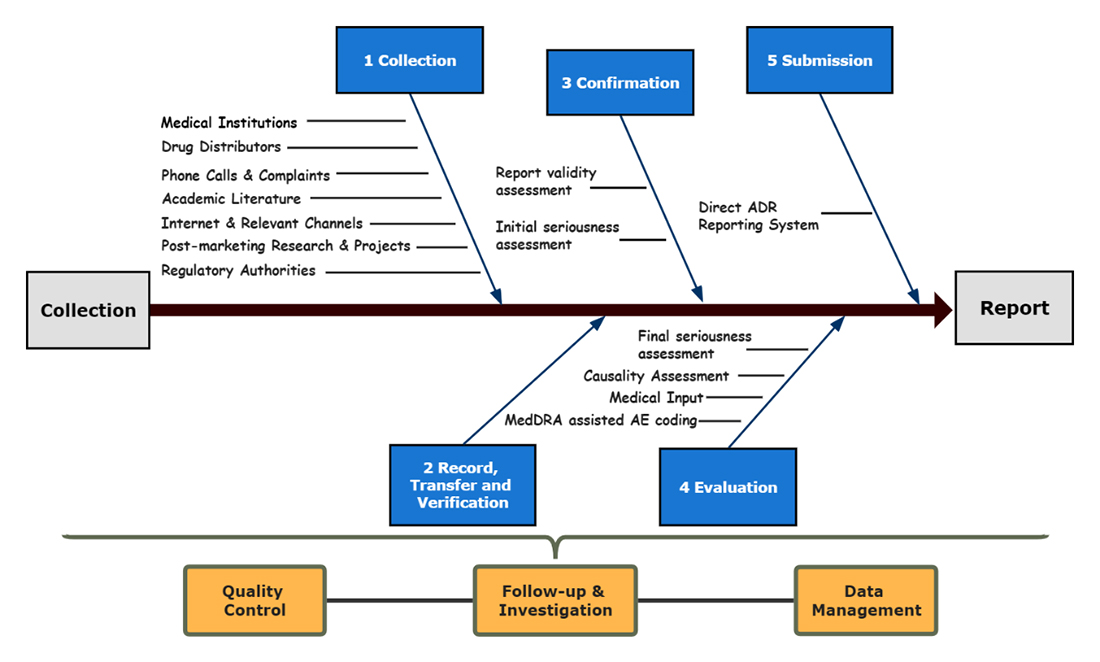
On December 19, 2018, China’s NMPA released the Guideline for Collecting and Reporting of Individual Case of Adverse Drug Reactions (hereinafter “the Guideline”). Aiming to standardize the collection and reporting throughout the Adverse Drug Reaction (hereinafter “ADR”) monitoring and processing, this Guideline provides for the Market Authorization Holder (MAH) a “5+3” instruction —5 consecutive procedures including: 1) “collection”, 2) “record, transfer, and verification”, 3) “confirmation”, 4) “evaluation”, and 5) “submission”; 3 important aspects throughout the process covering: 1) “quality control”, 2) “follow-up and investigation”, and 3) “data management”.
Accestra Consulting has digested the relevant regulations and compiled key points for you.
5 Consecutive Procedures
- Collection of Individual ADR Case
The MAH shall establish effective information channels for doctors, pharmacists, patients, and others, to actively collect ADR information related to clinical use, clinical studies, market projects, academic literature, etc. Effective channels often involve Medical Institutions, Drug Distributors, Hotline & Complaints, Academic Literature, Internet, Post-marketing Research & Projects, and Regulatory Authorities.- Medical Institutions
- The MAH may collect the clinical ADR information from the medical personnel regularly by means of daily visit, e-mail, telephone, fax, etc., make detailed records, and establish archives of ADR information.
- The MAH or its distributor shall make the medical institutions fully aware of the responsibility of reporting ADR and encourage medical personnel to report ADR when signing the drug purchase and sale contract with them.
- Drug Distributors
- The MAH shall establish an effective and direct channel for Drug Distributors to report ADR information.
- The MAH shall regularly evaluate the capability of the Distributor to perform its information collection responsibilities and take necessary measures to ensure the quantity and quality of the collected information.
- The MAH shall have a designated person to answer phone calls, collect and record ADR information reported by patients and others (e.g.,doctors, pharmacists, lawyers).
- The MAH shall inform the consumer in an effective manner of the ways to report ADR.
- The MAH shall report any ADR received through legal litigation channels, whether the report has been submitted to regulatory authorities by others or not.
- Academic Literature
- Academic literature is one of the high-quality sources to collect ADR information. The MAH shall research regularly and report the individual ADR cases found in academic literature.
- The literature screening shall be conducted at least every two weeks for new drugs first marketed or imported within 5 years, and once a month for other drugs in principle. Alternatively, the frequency could be adjusted according to the risk of the drug.
- The time frame of the searched literature shall be continuous as long as product is marketed.
- The MAH shall search on widely-used literature databases, including databases in China—such as China National Knowledge Infrastructure (CNKI), CQVIP database, Wanfang Data—and outside China like
PubMed, Embase, Ovid and others. - It is required to search at least two databases simultaneously in domestic and foreign literatures respectively.
- The MAH shall have a reasonable search strategy to ensure the comprehensiveness of search results and minimized omissions. For example, using the international nonproprietary name (INN)/active
ingredient of the drug as keywords for searching, or using a combination of generic names, trade names and other common names of the drug approved by the drug regulatory authorities.
- Internet and Relevant Channels
- The MAH shall regularly visit its websites—whether launched or managed—to collect potential ADR cases.
- The MAH shall collect ADR information from the company’s portal website, such as establishing a special report channel for ADR on the website, providing guidance on reporting pathways, report forms and report content, and publishing complete and up-to-date product instructions.
- Print media, digital media, social media/platforms under control of the MAH are also sources of individual ADR information, e.g., collection through corporate WeChat public accounts, microblogs, forums/bbs, etc.
- Post-marketing PV Research and Projects
- Individual ADR found in post-marketing studies initiated by enterprises (including studies abroad), or organized data collection programs shall be reported to regulatory authorities as required, such as clinical trials, non-interventional epidemiological studies, drug focus monitoring, patient support programs, market research or other marketing promotion projects.
- Chinese Regulatory Authorities
- The ADR report feedback from the domestic regulatory authorities to the MAH is mainly used for safety analysis and evaluation of the product.
- The MAH shall react to the feedback report within the required time limit, such as readjusting terminology, evaluating the seriousness and expectation, conducting relevance evaluation and others.
- Medical Institutions
- Record, Transfer, and Verification of Individual ADR Case
- Recording. The first person of MAH or its entrusting partner known of an individual ADR case is referred to as “the first recipient”.
- The first recipient shall try to obtain the ADR information as much as possible, including the patient’s condition, the reporter’s situation, the suspected combination use of drug, the occurrence of ADR.
- The ADR information received from various channels, such as emails, letters, phone calls, doctor interviews, etc., shall have original records.
- In addition to the reporter, other relevant persons providing the information of the case report shall be recorded as well to ensure that the information provider is identifiable
- The records shall be true, accurate and objective, and shall be kept properly
- Transferring. In the process of transferring the original records of individual ADR to the pharmacovigilance department by the first recipient, the authenticity and integrity of the records shall be maintained without deletion or omission. The transferring time shall be limited to ensure the timeliness of the report.
- Verifying. The MAH for China shall evaluate the authenticity and accuracy of the individual ADR information. And it shall try to verify the information when in doubt
- Confirmation of Individual ADR Case
- Contents to be confirmed mainly include: whether it is a valid report, whether it is within the scope of the report, whether it is a repeated report.
- 4 Elements of a valid report: A valid report should include the following 4 elements: 1) identifiable patients, 2) identifiable reporters, 3) suspected drugs, and 4) details of the ADR.
- If the 4 elements are incomplete, the report shall be deemed as invalid and shall be supplemented before reporting.
- The scope of the report includes:
a) the ADR occurring under the normal dosage of the drug;
b) the harmful reactions occurring under off-label drug use, such as off-indicated drug use, overdose, use with contraindications, etc.;
and
c) harmful reactions suspected to be caused by drug quality problems.
- Evaluation of Individual ADR Case in China
- After receiving individual ADR reports (including feedback reports from regulatory authorities), personnel responsible for pharmacovigilance activities shall evaluate the report, including judgement of new adverse reactions and serious adverse reactions, as well as evaluating the relevance between drugs and adverse reactions.
- The judgment of causality, also known as causality assessment, evaluates the correlation between the suspected drug and the adverse reaction/event of the patient.
- According to relevant guidelines by World Health Organization (WHO), causality assessment is classified into 6 terms—certain, probable/likely, possible, unlikely, conditional/unclassified, and unassessable/unclassifiable, see the table below:
Causality Term
If with time relationship to drug intake
If known
Response to withdrawal
Rechallenge
Other explanation
Certain + + + + – Probable/Likely + + + ? + Possible + ± ±? ? ±? Unlikely – – ±? ? ±? Conditional/Unclassified More data for proper assessment needed Unassessable/ Unclassifiable Cannot be judged because necessary information is insufficient
or unobtainable - Submission of Individual ADR Case
- The MAH in China shall submit individual ADR report via the direct adverse drug reaction reporting system— a platform supervised by China’s National Center for ADR Monitoring, and timely update and maintain the registration information in the system.
- The ADR reports shall be submitted within the following time limit:
a) Serious ADR in China shall be reported within 15 calendar days, and death cases shall be reported immediately.
b) Other non-serious ADR shall be reported within 30 calendar days.
c) Serious ADR abroad shall be reported within 15 calendar days.
3 Important Aspects
- Quality Control
- The MAH shall ensure that the contents of the report are true, complete, and accurate. The individual ADR shall be recorded in a true and detailed manner, and false reports are strictly prohibited.
- The names of the drug, the disease, the ADR, and the unit shall be filled in accurately and normatively.
- The names of the ADR as well as the disease, the diagnosis and the symptom shall be determined by referring to WHO Adverse Reactions Terminology (WHOART) or ICH Medical Dictionary for Regulatory Activities (MedDRA) and its supporting guidelines, such as MedDRA Term Selection: Points to Consider.
- Indicators of physical signs and laboratory test results shall be unbiased as the original records.
- Follow-up and investigation
- Follow-up. Individual ADR information received for the first time is generally incomplete. Thus, missing information should be followed up in the following priority order:
a) new and serious ADR cases
b) other serious ADR cases
c) new and non-serious ADR cases. - Investigation of cases of death. The MAH shall investigate known death cases and complete the investigation report and submit it within 15 calendar days. The patient’s medical records and autopsy report shall be collected according to the actual situation.
- The quality of the product shall also be reviewed during the investigation, and re-examined if necessary.
- Follow-up. Individual ADR information received for the first time is generally incomplete. Thus, missing information should be followed up in the following priority order:
- Data Management
- Data management shall run through the whole life cycle of the data. The management principles of truthfulness, integrity, safety, and traceability shall be adhered to from data collection, recording, transmission, processing, review, report, and storage to destruction.
- All electronic and paper data shall be kept with security and confidentiality.
- The ADR information of individual cases shall be well managed in the form of database for easy search, analysis, and evaluation, such as in Excel forms or a pharmacovigilance information system/platform of the MAH.
- The submitted ADR report forms shall be traceable to the original records, follow-up records and investigation reports.
- To implement strict access control to the database, the record of paper data shall be clear, readable, and understandable.
- Paper data shall be classified and catalogued for convenience of future search.
ADR Reporting is vital for patient safety and compliance in China. For advisory on complying with requirements and China’s GVP, Accestra Consulting is your professional and reliable partner.
Contact Us
For more information or request for support that tailored to your needs, please email: info@accestra.com





