

The new version of China’s “Drug Administration Law” has gone into effect since December 1st, 2019, which indicates a major step forward in China’s reformation of administration laws for drug products. Meanwhile, the National Medical Products Administration (NMPA) released the new version of “Provisions for Drug Registration” (hereinafter referred to as the New Provisions) and “Measures for Supervision and Administration of Drug Production” on March 30th, 2020. These regulations further consolidate all the new systems established by the “Drug Administration Law (Version 2019)”, such as the Marketing Authorization Holder (MAH) system, the Drug-related Associated Review & Approval system and the Priority Approval system, which provide the legal basis for drug registrations and transection structures of the pharmaceutical industry. This article aims to sort out the key points of the New Provisions which are relevant to the development, registration and transection of drug products, and could serve as reference for pharmaceutical enterprises.
1.Drug Registration System
1.1Changes of the Registration System
In the “Provisions for Drug Registration (Version 2007)” different application programs were set up for new drugs, generic drugs, imported drugs and OTC drugs respectively. Drug products received different approvals of registration based on their application programs, i.e. a new drug application obtained both a New Drug License and a Drug Approval No. while a generic drug application obtained only a Drug Approval No., and an imported drug application obtained a License of Imported Drug Registration (or a License of Medical Product Registration).
Registration rules identified by the New Provisions based on the new version of the “Drug Administration Law” are as follows:
Change 1 of Drug Registration: related contents of the New Drug License have been deleted (related concepts such as the New Drug License and the New Drug Monitoring Period are officially withdrawn).
Change 2 of Drug Registration: the registration approvals of new drugs, generic drugs and imported drugs have been unified as the License of Drug Registration.
Change 3 of Drug Registration: the concept of “imported drugs” is no longer available, as it has been substituted by the concept of “Drugs Manufactured Overseas” (the relevant rules are remained to be published).
Change 4 of Drug Registration: the market approval pathways of three drug categories have been identified as, 1) an integrated pathway of pre-clinical research to clinical trials to application for market approval; 2) a direct pathway of exempted clinical trials for generic drugs, in vitro diagnostic products administrated as drugs and other eligible products; 3) a direct pathway for Over the Counter Drugs (OTCs).
Change 5 of Drug Registration: the sub-packaging registration of drugs manufactured overseas is no longer applied at the Provincial Medical Products Administration (PMPA) and reviewed by the NMPA for the Drug Approval No., instead, simply filed at the PMPA.
1.2 Changes of Drug Classification
The New Provisions have combined and followed the rules in the previous version of “Provisions for Drug Registration” and “Reform Scheme for Registration Classification of Chemical Drugs” to classify drug registrations as follows:

Furthermore, NMPA have released the draft for public review for the detailed classification and corresponding requirements of application materials of TCMs, chemical drugs and biological products on April 30th, 2020.
2.Further Implementation of the MAH System
2.1 It’s clarified that MAH can be transferred
The Article 78 of the new Provisions clarify that the MAH transfer can be applied, approved and implemented by supplementary application pathway.
2.2 Make it clear that it’s mandatory to obtain production license for a MAH
The Article 50 of the new Provisions stipulate that the MAH shall obtain drug production license. Meanwhile, according to the relevant requirements in the new “Measures for Supervision and Administration of Drug Production”, it’s unnecessary for a MAH to have its own workshop, facility, equipment, hygienic environment and instruments, however it must be equipped with adequate technical staff as well as rules and standards which are complied with GMP guidelines.
3.Drug-related Associated Review & Approval
3.1 Introduction
The New Provisions continue to follow the drug-related associated review & approval system identified in the “CFDA Issued an Announcement on Adjusting the Evaluation and Approval for APIs, Excipients and Packaging Materials” (2017 No. 146). The detailed process is as follows:
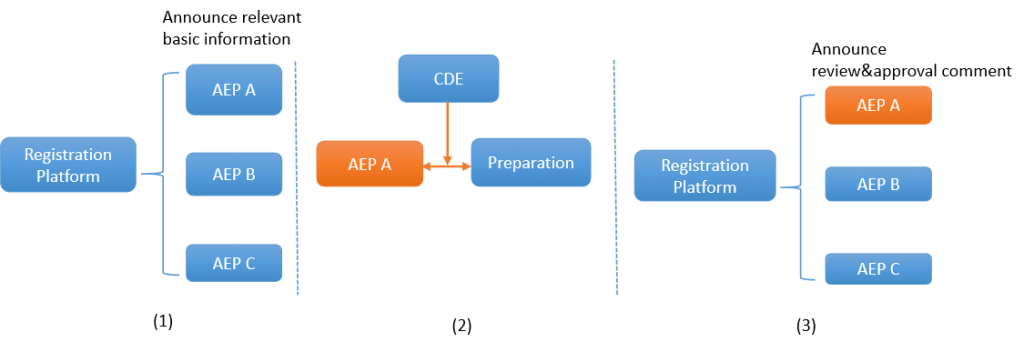
Pathway 1:
(1) After AEP (hereinafter referred to as AEP: APIs + excipients +packaging) production enterprises file their products on CDE’s platform, CDE will announce the basic information (such as product name, enterprise name, address, source and strength) for the selection of MAHs. (2) After a MAH chooses AEP A, CDE will conduct the review and approval of AEP A together with its associated drug product. (3) CDE will announce the final result of review & approval on the registration platform publicly.
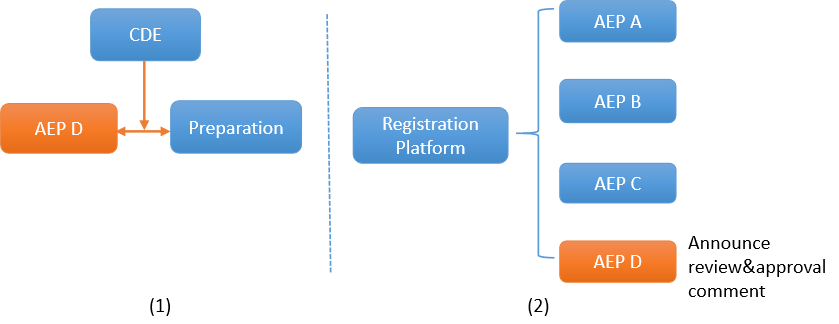
Pathway 2:
(1) A MAH can also choose an un-filed AEP D. However, it’s mandatory to submit relevant research materials of the AEP when applying for preparation registration. (2) CDE will announce the final result of review & approval on the registration platform publicly.
3.2 Implementation Status
Based on the data of CDE official database, until May 5th 2020, a total of 12,884 APIs are registered while 10,007 APIs are approved for their use in marketed FDF, including APIs produced overseas.
This article is summarized and translated from Jindu law firm.





