

To export a generic drug to China market, you need an Abbreviated New Drug Application (ANDA) for submission to China Center of Drug Evaluation (CDE) of National Medical Products Administration (NMPA formerly CFDA) for marketing authorization.
Highlights of China’s New Regulations for ANDA Submission
The requirements for China ANDA submission have changed and improved over recent years. These changes reduce complexity, shorten timelines, lower budget costs, and align closely with the international community. Highlights include:
- Conditional acceptance of foreign clinical data,
- Fast track review pathway,
- Simplification of bioequivalence (BE) study application,
- Adoption of ICH M4 CTD format for drug dossier.
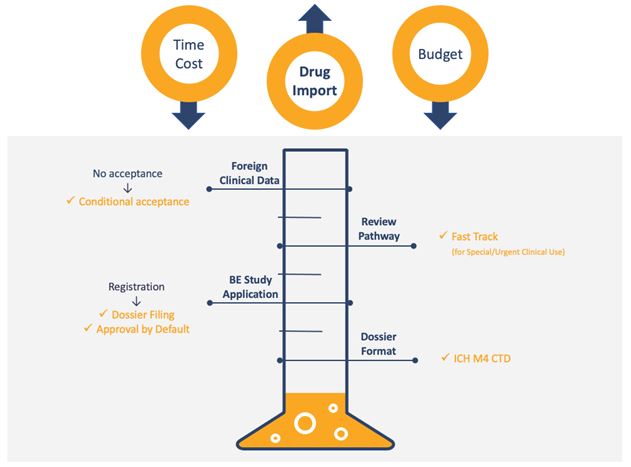
Fig.1 Regulatory Reform and Impact on Generic Drug Import Market
TERM: BE Bioequivalence; ICH The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; CTD Common Techincal Document
Who can apply for an ANDA in China?
According to the latest Provisions for Drug Registration (2020), a foreign applicant needs to choose a China domestic legal entity as the local representative to apply for and submit ANDA.
The foreign applicant can either choose to establish a legal entity or branch in China or appoint a qualified local agent as a representative. You can contact Accestra Consulting for local agent representation services.
Generic Drug Development Requirements
Reference Listed Drug (RLD)
China CDE requires that the reference product a generic drug refers to is on CDE’s RLD List. If you are submitting an ANDA while the reference product is not on the List, a RLD application will be required.
Clinical Trial/BE Study
China NMPA issued several guidelines for generics of different dosage forms and administration routes to assist applicants identify whether their drugs can be completely exempted from clinical studies.
If your drug needs clinical studies and you have finished them outside of China, your clinical data could be accepted by CDE, and additional clinical trial/BE study could be exempted in China.
According to CDE’s review of the drug dossier, there are four different scenarios for an imported generic where clinical studies are exempted or required:
Table 1. Clinical Study Requirements of An Imported Generic Drug
|
NO CLINICAL STUDIES REQUIRED IN CHINA |
||
|---|---|---|
| Clinical study exemption | ||
|
CLINICAL STUDIES REQUIRED IN CHINA |
||
| Foreign clinical data available | Data fully accepted | Clinical study exemption |
| Data partly accepted/rejected | Clinical trial*/BE study required | |
| No foreign clinical data | Clinical trial*/BE study required |
Clinical trial*: Certain clinical trial is required in China, incl. bridging study, confirmatory phase III clinical trial, and others.
ANDA Submission Requirements
The ANDA review and approval process are as below:
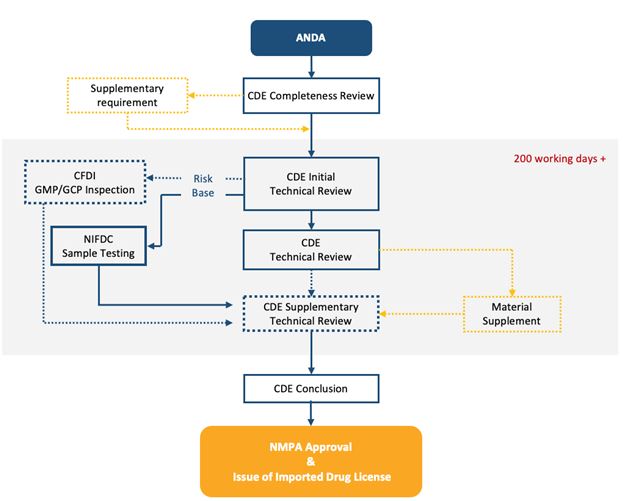
Fig.2 NMPA Workflow ANDA Review and Approval
TERM: ANDA abbreviated new drug application; NMPA National Medical products Administration; CDE Center for Drug Evaluation; CFDI Center for Food and Drug Inspection; NIFDC National Institutes for Food and Drug Control
TERM: ANDA abbreviated new drug application; NMPA National Medical products Administration; CDE Center for Drug Evaluation; CFDI Center for Food and Drug Inspection; NIFDC National Institutes for Food and Drug Control
Timeline
The overall timeline from ANDA submission to approval can take approximately 12-16 months.
CDE’s technical review part is around 200 working days, which could be extended to another 1/3 if material supplementation is required.
Costs
- The administrative costs are approximately $80,000 if clinical studies have been or will be conducted,
- or $60,000 if clinical studies are completely exempted.
Fast Track Review Pathway
A generic drug is reviewed by fast track if CDE determines that it could meet the market shortage or treat a rare or children’s disease.
Regardless of whether the drug has been market authorized in the US or EU, the applicant can apply for the fast track review pathway in China to speed up drug approval.
Our Tips
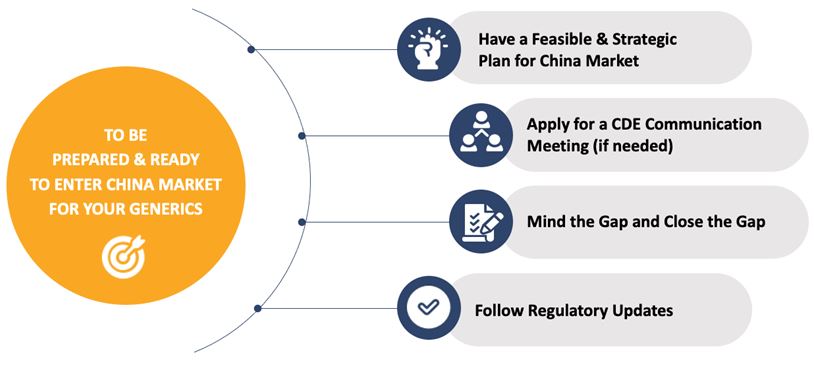
Fig.3 Activities an ANDA Application can do before Application.
TERM: CDE Center for Drug Evaluation1. Have a Feasible & Strategic Plan for China Market
Make a feasible and strategic plan for ANDA submission and explore the most appropriate pathway options to China.
It is important to understand the pros and cons of different routes to market authorization.
A good market access strategy and implementation plan could not only save a significant amount of time and cost but most importantly reduce risks and increase the chance of success.
2. Apply for a CDE Communication Meeting (if needed)
If CDE’s requirements for your product are not explicit, a communication meeting can be applied to know CDE’s initial opinions on any critical issues relating to CMC (Chemical, Manufacturing, and Control), non-clinical and clinical studies, and to shorten the application timeline.
3. Mind the Gap and Close the Gap
After the regulatory reforms of ANDA submission, China regulations are now more aligned with the international requirements and ICH guidelines.
Except for the similarities, differences are still present between China, EU, and the US systems, especially in CTD Module 1 and CMC requirements .
It is important to conduct data gap analysis to identify and close gaps under China regulations.
4. Follow Regulatory Updates
China’s drug policy is young and significant changes happen frequently, so make sure to keep up with the regulatory updates.
Accestra Consulting can guide and assist you in fulfilling all the regulatory requirements for your generic drug product before, during, and after ANDA, including but not limited to the following services:
- Authorized local agent,
- Regulatory strategy plan,
- RLD confirmation & application,
- CDE communication meeting,
- Data gap analysis,
- ANDA dossier preparation, submission management & CDE review follow-up,
- Annual report,
- Pharmacovigilance (PV).





