

Background
It has been almost 13 years since the implementation of China’s “Provisions for Drug Registration” (SFDA Order No.28) on Oct 1st, 2007. Over the past ten years, these provisions have played a very important role in ensuring drug’s safety, efficacy and controllable quality, and regulating behaviors in drug registration. But with the continuous improvement of global harmonization efforts around the world and the need for reform and development of China’s pharmaceutical industry, this was followed by the beginning of groundbreaking promulgation of “Review & Approval System of Drugs and Medical Device” (No. 44 [2015] by the State Council). Against this background, the Order No.28 implemented in 2007 could not meet the requirements of drugs’ registration application and review under the new situation. Based on the “Drug Administration Law” issued and implemented on Dec 1st, 2019, the new “Provisions for Drug Registration” has has finally been implemented after 4 months. This clarification is a significant market access milestone for international pharmaceuticals community.
This article mainly illustrates the issuance and implementation of new version of “Provisions for Drug Registration” (hereinafter referred to as “Provisions”) has multiple obvious effects on the applications of FDF (finished dosage form) products.
Q1: The interpretation of MAH (Market Authorization Holder) system in “Provisions”
A: The current “Drug Administration Law” introduce the “MAH” chapter for the first time, and stipulate the definition, responsibility, right and obligation of MAH in detail. The Article 3 in “Provisions” writes that “After obtaining drug registration certification, the applicant could be a MAH”. And the following chapter also illustrates the responsibility and obligation of MAH in the administration of drug registration.
Q2: With the post-implementation of “Provisions”, the overseas applicants (applicants of FDF products) of China could also be MAH. Then what are the conditions overseas applicants shall have simultaneously?
A: It is clearly stated in Article 38 of Chapter 3 of “Drug Administration Law” that if the MAH is an overseas enterprise, this enterprise shall appoint its legal person in China perform the obligations of MAH and bear joint responsibilities.
Q3: What is the impact of the new classification on the registration work, as drug registration is managed according to the classification of traditional Chinese medicine, chemical drugs and biological products?
A: The “Provisions” unifies the classification standard of traditional Chinese medicine, chemical drug and biological product, which means they are classified to innovative new drugs, improved new drugs and generic drugs by innovation degree. And different generic products have different name of its category as the table below shows.
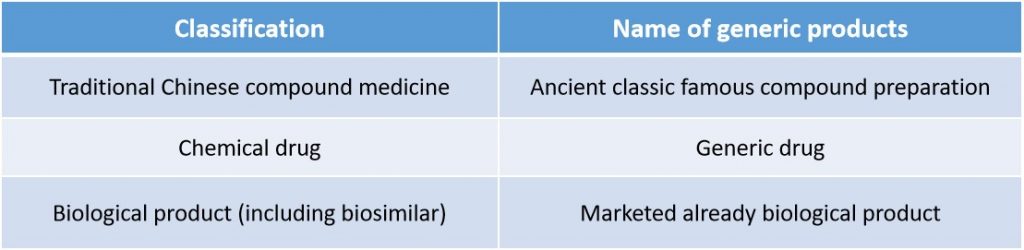
At the same time, the “Provisions” mentions that the detailed classification and corresponding requirements for application materials will be formulated and issued by NMPA in accordance with product characteristic, innovation degree of drug and management need of review&approval. As for the application of chemical drug, NMPA will update the announcement on the work plan for the reform of the classification of chemical drug registration (namely No.51, 2016). We’ll continue to focus on what the specific writing requirements for registration materials of each category.
Q4: Is there any difference between the clinical trial negative approval timeline in the “Provisions”, that applications for clinical trials shall be decided whether or not within 60wd from the date of acceptance, and that of No.50, 2018? And what is the impact of this difference on the real registration application?
A: Here is basically the same as “Announcement of NMPA on Adjusting the Review&Approval Procedure of Drug Clinical Trials” (No.50, 2018) promulgated by NMPA on July 7th, 2018. However, the details have some obvious changes. For example, the timeline of clinical trial mentioned in No.50, 2018 is “Within 60wd from the date of application acceptance and payment, while “Applications for clinical trials shall be decided whether or not within 60wd from the date of acceptance” in the “Provisions”. Especially for 80% of imported FDF items, it may take almost 2 months for an applicant to complete the step of payment, as the several rounds of communication between CDE and applicant for exchange rate and influences of domestic and foreign holidays. “Provisions” will really benefits overseas applicants, if it is implemented and counted time from acceptance.
Q5: What is the significance of proposing the safety update report during the development for the clinical trial study?
A: This item echoes the negative approval system of 60wd in “Announcement on Adjusting the Review&Approval Procedure for Clinical Trials of Medical Device”, which has provided definite evidence for applicants’ standard operation during clinical trials.
Q6: What is the specific impact of the implementation of BE study filing system and promulgation of “Technical Guidance for the Acceptance of Overseas Clinical Trial Data of Drug” on drug registration work?
A: This item corresponds perfectly to the No.257, 2015, that is “Announcement on Management of Implementing BE Study Filing System of Chemical Drugs by CFDA”, and provides a very good policy entry for the registration of generic drugs. The long process of “Applying for clinical trial” is not a mandatory requirement for oral solid generic drug applicant to deploy BE study. The applicant can conduct BE study after BE filing according to current regulations, which not only greatly saves power of review but also shortens the timeline of application of generic drugs.
In addition, the implementation of the two policies is very favorable for the application of imported oral solid preparations. With the promulgation of “Technical Guidance for the Acceptance of Overseas Clinical Trial Data of Drug” by NMPA on July 6th , 2018, imported oral solid preparation applicants can apply for market approval directly by using the BE study data that has been completed and approved abroad. The applicants have more advantages in obtaining review&approval in advance compared with the application of domestic generic drugs. Herein please note that the quality of overseas clinical trial must meet the relevant requirements of regulations and laws in China. For instance, an overseas applicant has only conducted fasting BE study when applying for EMA approval. But according to the domestic requirements of regulations and laws, it’s mandatory to provide both fasting and fed BE study data. As a result, in order to obtain the final approval, this overseas applicant shall supplement fed BE study with the availability of BE study data.
Q7: What is the influence of the implementation of drug-related associated review&approval system of API, pharmaceutical excipients and packaging materials (hereinafter referred to as AEP) on FDF registration application? What are the contents FDF applicants shall take in consideration?
A: The implementation of FDF review and drug-related associated review&approval strengthens FDF applicants’ main responsibility, and reminds FDF applicants that they shall consider from all aspects when choosing supplier of AEP. FDF applicants shall not only consider whether the quality of their AEP meet the process of FDF products, but also confirm whether the enterprise of AEP is willing to cooperate with FDF application to register on CDE platform and other possible inspections. Since these factors are related to whether the FDF products could be approved successfully.
Q8: The Article 42 of “Provisions” stipulate that if FDF applicant choose the AEP has not been registered, the relevant materials of AEP shall be applied together with FDF registration. What are the main effects of this provision on the acceptance link of FDF products application?
A: The spotlight of this item is the effect on acceptance link. For applicants especially for overseas applicants, it is not necessary for them to communicate repeatedly with supplier of AEP, and persuade the supplier to file on the CDE platform. According to the acceptance policy implemented about half a year ago, if the eligible AEP does not file on the platform, FDF applicant is required to provide an exclusive statement in the certification documents section and submit the full set of materials of AEP in the preparation documents when submitting the application for registration. Which means no exclusive statement will not be accepted. Actually AEP suppliers always provide their goods simultaneously to multiple FDF enterprises. In that case, it will be a fake statement if provided. Recently, with the adjustment of acceptance policy and the promulgation of “Provisions”, applicants and their AEP suppliers will no longer face this kind of embarrassing situation.
This article is originally translated from Canny.
Glossary
FDF Finished Dosage Form
MAH Marketing Authorization Holder
AEP API, Pharmaceutical Excipients and Packaging Materials





